
03.5 Análisis de microarrays¶
Objetivos de la práctica
Utilizar datos de microarrays para agrupar genes en función de sus patrones de expresión.
Comparar los grupos obtenidos con los diferentes tipos de cáncer anotados.
El conjunto de datos NCI60 contiene información genética de 64 líneas celulares cancerígenas. Para cada una de ellas, se ha cuantificado la expresión de 6830 genes mediante tecnología microarray. Se conoce el tipo de cáncer (histopatología) al que pertenece cada línea celular y se puede utilizar esta información para evaluar si el método de clustering (agglomerative hierarchical clustering) es capaz de agrupar correctamente las líneas empleando los niveles de expresión génica.
import matplotlib.pyplot as plt
%matplotlib inline
import pandas as pd
import numpy as np
from skimage import io
from IPython import display
from bokeh.io import output_notebook, show
from bokeh.plotting import figure
output_notebook()
Carga de Datos¶
Descargar el archivo que se encuentra en Moodle con el nombre nci_data.csv y situarlo en una carpeta de nombre data en el path donde se encuentre el cuaderno Jupyter.
df = pd.read_csv('data/nci_data.csv')
df.head()
| Gene | s1 | s2 | s3 | s4 | s5 | s6 | s7 | s8 | s9 | ... | s55 | s56 | s57 | s58 | s59 | s60 | s61 | s62 | s63 | s64 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | g1 | 0.300 | 0.679961 | 0.940 | 0.280 | 0.485 | 0.310 | -0.830 | -0.190 | 0.460 | ... | 0.010 | -0.620 | -0.380 | 0.04998 | 0.650 | -0.030 | -0.270 | 0.210 | -0.050 | 0.350 |
| 1 | g2 | 1.180 | 1.289961 | -0.040 | -0.310 | -0.465 | -0.030 | 0.000 | -0.870 | 0.000 | ... | -1.280 | -0.130 | 0.000 | -0.72002 | 0.640 | -0.480 | 0.630 | -0.620 | 0.140 | -0.270 |
| 2 | g3 | 0.550 | 0.169961 | -0.170 | 0.680 | 0.395 | -0.100 | 0.130 | -0.450 | 1.150 | ... | -0.770 | 0.200 | -0.060 | 0.41998 | 0.150 | 0.070 | -0.100 | -0.150 | -0.090 | 0.020 |
| 3 | g4 | 1.140 | 0.379961 | -0.040 | -0.810 | 0.905 | -0.460 | -1.630 | 0.080 | -1.400 | ... | 0.940 | -1.410 | 0.800 | 0.92998 | -1.970 | -0.700 | 1.100 | -1.330 | -1.260 | -1.230 |
| 4 | g5 | -0.265 | 0.464961 | -0.605 | 0.625 | 0.200 | -0.205 | 0.075 | 0.005 | -0.005 | ... | -0.015 | 1.585 | -0.115 | -0.09502 | -0.065 | -0.195 | 1.045 | 0.045 | 0.045 | -0.715 |
5 rows × 65 columns
Los 64 tipos de cancer se encuentra en el archivo nci_label.txt también situado en Moodle y se descarga a la misma carpeta data
df_lbl = pd.read_csv('data/nci_label.txt', header=None, names=["Cell"])
df_lbl.head()
labels_ok = df_lbl.values
np.size(labels_ok), np.unique(labels_ok)
(64,
array(['BREAST', 'CNS', 'COLON', 'K562A-repro', 'K562B-repro', 'LEUKEMIA',
'MCF7A-repro', 'MCF7D-repro', 'MELANOMA', 'NSCLC', 'OVARIAN',
'PROSTATE', 'RENAL', 'UNKNOWN'], dtype=object))
La estructura del fichero nci_data.csv es:
Una columna que sirve para enumerar las 6830 expresiones genéticas y que no se utilizará.
64 columnas con los tipos de cancer.
Por tanto, se toman las 64 últimas columnas, por lo que es necesario transponer la matriz X_trap para obtener el conjunto inicial de entrenamiento, que se denomina X_ini.
X_ini ya tiene 64 filas por cada tipo histológico de cancer con 3680 características, siendo cada uno de ellos la expresión de un gen.
X_trap = df.values[:,1:65]
X_ini = np.transpose(X_trap)
X_ini.shape
(64, 6830)
Estandarización de los datos de entrada
Para poder probar con datos estandarizados o no estandarizados en la siguiente celda tenemos las dos opciones de forma que en la matriz X con la que se va a trabajar se podrá volcar el dato estandarizado o no, según se comente o descomente la última línea de código
from sklearn.preprocessing import StandardScaler
scaler = StandardScaler()
X = scaler.fit_transform(X_ini)
#X = X_ini
Elección del número de clusters¶
Criterio de Inercia (suma de distancias al cuadrado a los centroides)¶
Se calcula la inercia relativa a Suma de Distancias al Cuadrado del modelo completo, que se puede calcular con 1 cluster
from sklearn.cluster import KMeans
import sklearn.metrics as metrics
km = KMeans(n_clusters=1, init='k-means++', max_iter=300, tol=1e-04, random_state=1)
km.fit(X)
tss=km.inertia_
distortions = []
for i in range(2, 20):
km = KMeans(n_clusters=i, init='k-means++', max_iter=300, tol=1e-04)
km.fit(X)
distortions.append(km.inertia_/tss)
p = figure(width=700, plot_height=400)
p.line(range(2,20), distortions, line_color="navy", line_width=3, line_alpha=0.6, legend_label="Inertia/tss vs k")
p.legend.location='top_right'
p.legend.click_policy="hide"
p.xaxis.axis_label = 'Número de Clusters'
p.yaxis.axis_label = 'Inertia/TSS'
show(p)
Conclusión del gráfico del codo: no resulta muy concluyente por el escaso número de registros de la matriz X, por lo que se echa mano de otro criterio de validación intrínseca que es el criterio de la Silueta
Criterio de la Silueta¶
El coeficiente de silueta se calcula utilizando la distancia media dentro del grupo (a) y la distancia media del grupo más cercano (b) para cada muestra. El Coeficiente de silueta para una muestra es (b - a) / max(a, b). Para aclarar, b es la distancia entre una muestra y el grupo más cercano del que la muestra no forma parte. El coeficiente de silueta solo se define si el número de etiquetas es \(2 \ge N_{labels} \ge N_{samples} - 1\)
El mejor valor es 1 y el peor valor es -1. Los valores cercanos a 0 indican clústeres superpuestos. Los valores negativos generalmente indican que una muestra se ha asignado al conglomerado equivocado, ya que un conglomerado diferente es más similar.
from sklearn.cluster import KMeans
import sklearn.metrics as metrics
siluetas = []
for i in range(2, 20):
km = KMeans(n_clusters=i, init='k-means++', max_iter=300, tol=1e-04, random_state=1)
km.fit(X)
silueta = metrics.silhouette_score(X,km.labels_,metric="euclidean",sample_size=1000)
#print(i, km.inertia_, tss, km.inertia_/tss)
siluetas.append(silueta)
p = figure(width=500, plot_height=300)
p.line(range(2,20), siluetas, line_color="navy", line_width=3, line_alpha=0.6)
p.legend.location='top_left'
p.legend.click_policy="hide"
p.xaxis.axis_label = 'Número de Clusters'
p.yaxis.axis_label = 'Silueta'
show(p)
/usr/local/lib/python3.8/dist-packages/bokeh/models/plots.py:766: UserWarning:
You are attempting to set `plot.legend.location` on a plot that has zero legends added, this will have no effect.
Before legend properties can be set, you must add a Legend explicitly, or call a glyph method with a legend parameter set.
warnings.warn(_LEGEND_EMPTY_WARNING % attr)
/usr/local/lib/python3.8/dist-packages/bokeh/models/plots.py:766: UserWarning:
You are attempting to set `plot.legend.click_policy` on a plot that has zero legends added, this will have no effect.
Before legend properties can be set, you must add a Legend explicitly, or call a glyph method with a legend parameter set.
warnings.warn(_LEGEND_EMPTY_WARNING % attr)
Conclusión del gráfico de la Silueta: Los mejores valores están primero en 8 y luego en 16 grupos.
Vamos a crear una variable con el número de grupos para poder cambiar dicho valor sin tocar el resto de código
Como se sabe que hay 14 tipos de cancer, este debería ser el valor teórico que nos debería haber salido si el clustering ajustara perfectamente la realidad
print("Total tipos de cancer", np.size(np.unique(labels_ok)))
totalClusters = 14
Total tipos de cancer 14
Cluster Jerarquico¶
Se calcula con el número de clusters indicado anteriormente
from sklearn.cluster import AgglomerativeClustering
ac = AgglomerativeClustering(n_clusters=totalClusters, affinity='euclidean', linkage='complete', compute_distances=True)
#ac = AgglomerativeClustering(n_clusters=14, affinity='euclidean', linkage='complete', distance_threshold=None)
y_ag = ac.fit_predict(X)
y_ag, ac.distances_[-1], 0.7*ac.distances_[-1], ac.n_clusters_
(array([ 2, 2, 2, 9, 1, 1, 1, 1, 0, 2, 2, 2, 2, 2, 6, 6, 6,
4, 4, 8, 6, 6, 0, 0, 0, 0, 0, 6, 0, 0, 2, 0, 0, 3,
3, 3, 3, 3, 13, 13, 12, 0, 10, 0, 10, 10, 10, 10, 5, 5, 5,
5, 0, 0, 11, 7, 7, 7, 9, 7, 7, 7, 7, 7]),
163.48974041519423,
114.44281829063596,
14)
Se echa un primer vistazo al aspecto que tiene la agrupación conseguida
etiquetas=np.unique(y_ag)
numEtiquetas = np.size(etiquetas)
maxSizeCluster=0
for et in etiquetas:
print(et, np.size(y_ag[y_ag==et]), y_ag[y_ag==et])
0 14 [0 0 0 0 0 0 0 0 0 0 0 0 0 0]
1 4 [1 1 1 1]
2 9 [2 2 2 2 2 2 2 2 2]
3 5 [3 3 3 3 3]
4 2 [4 4]
5 4 [5 5 5 5]
6 6 [6 6 6 6 6 6]
7 8 [7 7 7 7 7 7 7 7]
8 1 [8]
9 2 [9 9]
10 5 [10 10 10 10 10]
11 1 [11]
12 1 [12]
13 2 [13 13]
Se lista con mayor detalle los grupos obtenidos y los tipos de cancer asignados a cada uno de ellos con su nombre
pd.set_option('display.width', 1000)
tiposCancerGrupo = ['' for i in range(numEtiquetas)]
for iCancer in range(np.size(y_ag)):
iCluster = y_ag[iCancer]
tiposCancerGrupo[iCluster] += str(labels_ok[iCancer]) + ";"
for i in range(np.size(tiposCancerGrupo)):
print(i, tiposCancerGrupo[i])
0 ['NSCLC'];['MELANOMA'];['PROSTATE'];['OVARIAN'];['OVARIAN'];['OVARIAN'];['OVARIAN'];['PROSTATE'];['NSCLC'];['NSCLC'];['COLON'];['COLON'];['NSCLC'];['NSCLC'];
1 ['BREAST'];['CNS'];['CNS'];['BREAST'];
2 ['CNS'];['CNS'];['CNS'];['NSCLC'];['RENAL'];['RENAL'];['RENAL'];['RENAL'];['NSCLC'];
3 ['LEUKEMIA'];['K562B-repro'];['K562A-repro'];['LEUKEMIA'];['LEUKEMIA'];
4 ['BREAST'];['NSCLC'];
5 ['MCF7A-repro'];['BREAST'];['MCF7D-repro'];['BREAST'];
6 ['RENAL'];['RENAL'];['RENAL'];['UNKNOWN'];['OVARIAN'];['OVARIAN'];
7 ['MELANOMA'];['BREAST'];['BREAST'];['MELANOMA'];['MELANOMA'];['MELANOMA'];['MELANOMA'];['MELANOMA'];
8 ['RENAL'];
9 ['RENAL'];['MELANOMA'];
10 ['COLON'];['COLON'];['COLON'];['COLON'];['COLON'];
11 ['NSCLC'];
12 ['LEUKEMIA'];
13 ['LEUKEMIA'];['LEUKEMIA'];
Función a la medida para graficar el Dendograma
Utiliza la paleta de colores básica de pyplot:
[‘b’, ‘g’, ‘r’, ‘c’, ‘m’, ‘y’] : Azul, Verde, Rojo, Cian, Magenta, Amarillo
En la llamada a la función se reserva el color ‘k’ (Negro) para los puntos aislados en el dendograma. Esto se realiza con la variable above_threshold_color.
Para mejorar la visión se ajusta el tamaño del fuente (leaf_font_size) a 10
La función scipy.cluster.hierarchy.dendrogram a la que se llama en plot_dendrogram tiene un parámetro opcional que es color_threshold. Este parámetro le sirve para situar la linea de corte de grupos en el dendograma. Por debajo de esta distancia crea grupos con un color distinto cada uno, salvo que estén aislados.
Por defecto, si no se pasa ningún valor, toma 0.7 * Altura_maxima . Se incorpara código a plot_programa para calcular automáticamente la altura umbral en función de los grupos formados en el cluster
import numpy as np
from matplotlib import pyplot as plt
from scipy.cluster.hierarchy import dendrogram
from sklearn.datasets import load_iris
from sklearn.cluster import AgglomerativeClustering
from scipy.cluster.hierarchy import set_link_color_palette
def plot_dendrogram(model, subtitulo, **kwargs):
# Create linkage matrix and then plot the dendrogram
##Busqueda automática de la distancia umbral
nSamples = len(model.labels_)
maxChild = max(max(model.children_[:,0]),max(model.children_[:,1]))+1
etiquetaNodo=[-1 for i in range(maxChild)]
distBajoUmbral=[]
for i, nodo in enumerate(model.children_):
etiq1 = model.labels_[nodo[0]] if nodo[0] < nSamples else etiquetaNodo[nodo[0]]
etiq2 = model.labels_[nodo[1]] if nodo[1] < nSamples else etiquetaNodo[nodo[1]]
if etiq1 == etiq2 and etiq1>-1:
etiquetaNodo[i+nSamples]=etiq1
distBajoUmbral.append(model.distances_[i])
distanciaUmbral = 0 if distBajoUmbral==[] else max(distBajoUmbral)
if distanciaUmbral > 0: ## La distancia umbral ha de queda en punto medio valor calculado más distancia siguiente nodo
distanciaUmbral = 0.5*(distanciaUmbral + min(model.distances_[model.distances_ > distanciaUmbral]))
if 'color_threshold' not in kwargs.keys(): ## Si el valor no viene por parámetro se añade
kwargs['color_threshold'] = distanciaUmbral
# create the counts of samples under each node
counts = np.zeros(model.children_.shape[0])
n_samples = len(model.labels_)
for i, merge in enumerate(model.children_):
current_count = 0
for child_idx in merge:
if child_idx < n_samples:
current_count += 1 # leaf node
else:
current_count += counts[child_idx - n_samples]
counts[i] = current_count
linkage_matrix = np.column_stack([model.children_, model.distances_,
counts]).astype(float)
# Plot the corresponding dendrogram
#dendrogram(linkage_matrix, **kwargs)
# Plot the corresponding dendrogram
fig, ax = plt.subplots(1, 1, figsize=(15, 10)) # set size
fig.suptitle("Dendograma de Clustering Jerárquico")
set_link_color_palette(['b', 'g', 'r', 'c', 'm', 'y'])
if 'above_threshold_color' not in kwargs.keys(): ## Si el valor no viene por parámetro se añade
kwargs['above_threshold_color'] = 'k'
ax = dendrogram(linkage_matrix, **kwargs)
plt.tick_params(axis='x', bottom='off', top='off', labelbottom='off')
plt.tight_layout()
plt.xlabel(subtitulo)
### Se añade una barra con el umbral
w_c = kwargs['above_threshold_color']
#w_y = 0.7*model.distances_[-1] if 'color_threshold' not in kwargs.keys() else kwargs['color_threshold']
w_y = kwargs['color_threshold']
if w_y > 0:
xmin, xmax, ymin, ymax = plt.axis()
plt.plot([w_y for n in range(int(xmax-xmin))], c=w_c, ls='--')
plt.show()
# , color_threshold=125 : Para corte por nivel de distancia umbral
## Por defecto el toma 0,7 de la distancia máxima como valor del parámetro "color_threshold"
plot_dendrogram(ac, "Tipos de cancer", labels=labels_ok, leaf_font_size=10)
#plot_dendrogram(ac, "Tipos de cancer", labels=labels_ok, above_threshold_color='k', leaf_font_size=10, color_threshold=115)
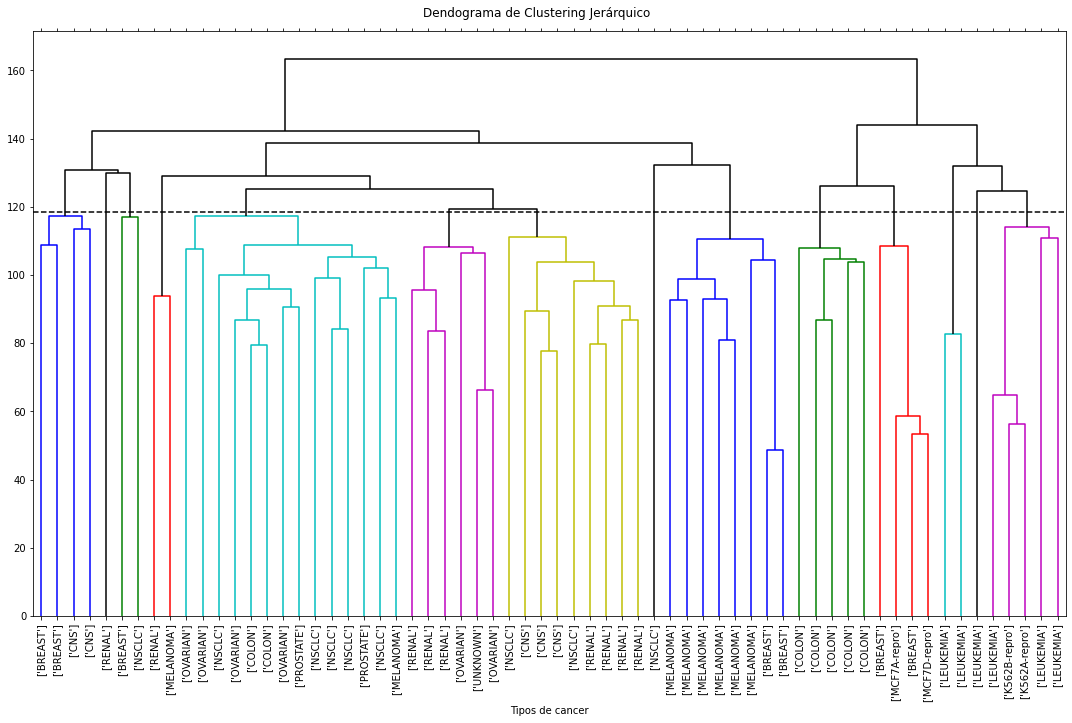
Se valida el cluster jerárquico¶
ac = AgglomerativeClustering(n_clusters=14, affinity='euclidean', linkage='complete')
y_ag = ac.fit_predict(X)
from sklearn.metrics.cluster import rand_score
from sklearn.metrics.cluster import adjusted_rand_score
y_ok = [lbl[0] for lbl in labels_ok]
## Rand - Puntuación de similitud entre 0.0 y 1.0, (1.0 significa coincidencia perfecta)
R=rand_score(labels_true=y_ok, labels_pred=y_ag)
ARI=adjusted_rand_score(labels_true=y_ok, labels_pred=y_ag)
R, ARI, y_ok
(0.8720238095238095,
0.2645020065669464,
['CNS',
'CNS',
'CNS',
'RENAL',
'BREAST',
'CNS',
'CNS',
'BREAST',
'NSCLC',
'NSCLC',
'RENAL',
'RENAL',
'RENAL',
'RENAL',
'RENAL',
'RENAL',
'RENAL',
'BREAST',
'NSCLC',
'RENAL',
'UNKNOWN',
'OVARIAN',
'MELANOMA',
'PROSTATE',
'OVARIAN',
'OVARIAN',
'OVARIAN',
'OVARIAN',
'OVARIAN',
'PROSTATE',
'NSCLC',
'NSCLC',
'NSCLC',
'LEUKEMIA',
'K562B-repro',
'K562A-repro',
'LEUKEMIA',
'LEUKEMIA',
'LEUKEMIA',
'LEUKEMIA',
'LEUKEMIA',
'COLON',
'COLON',
'COLON',
'COLON',
'COLON',
'COLON',
'COLON',
'MCF7A-repro',
'BREAST',
'MCF7D-repro',
'BREAST',
'NSCLC',
'NSCLC',
'NSCLC',
'MELANOMA',
'BREAST',
'BREAST',
'MELANOMA',
'MELANOMA',
'MELANOMA',
'MELANOMA',
'MELANOMA',
'MELANOMA'])
from sklearn.metrics.cluster import mutual_info_score
from sklearn.metrics.cluster import adjusted_mutual_info_score
VI = mutual_info_score(labels_true=y_ok, labels_pred=y_ag)
## El adj_VI devuelve un valor de 1 cuando las dos particiones son idénticas (es decir, coinciden perfectamente)
## Las particiones aleatorias (etiquetados independientes) tienen una AMI esperada de alrededor de 0 en promedio,
#por lo que pueden ser negativas
adj_VI = adjusted_mutual_info_score(labels_true=y_ok, labels_pred=y_ag)
VI, adj_VI
(1.5102113060840772, 0.4099076587645776)